You find a single hair at a crime scene. To most people, it’s just hair. To a forensic scientist, it is a potential key witness. But before that tiny strand can testify in court, it has to go through a rigorous chemical journey. This journey is called DNA extraction, which is the process of isolating and purifying deoxyribonucleic acid from biological cells for analysis. It is the critical first step in forensics, medical diagnostics, and genetic research. Without clean DNA, every subsequent test fails.
Think of DNA extraction like trying to pull a specific thread out of a tangled sweater without breaking it. You have to remove the fabric (cell membranes), the dye (proteins), and the lint (other cellular debris) to get just the thread (DNA). The goal is always the same: separate the DNA from everything else so it is pure enough for downstream applications like polymerase chain reaction (PCR) or sequencing.
The Universal Four-Step Framework
No matter what kit you buy or what lab protocol you follow, DNA extraction always follows a logical four-step order. Understanding this framework helps you troubleshoot when things go wrong.
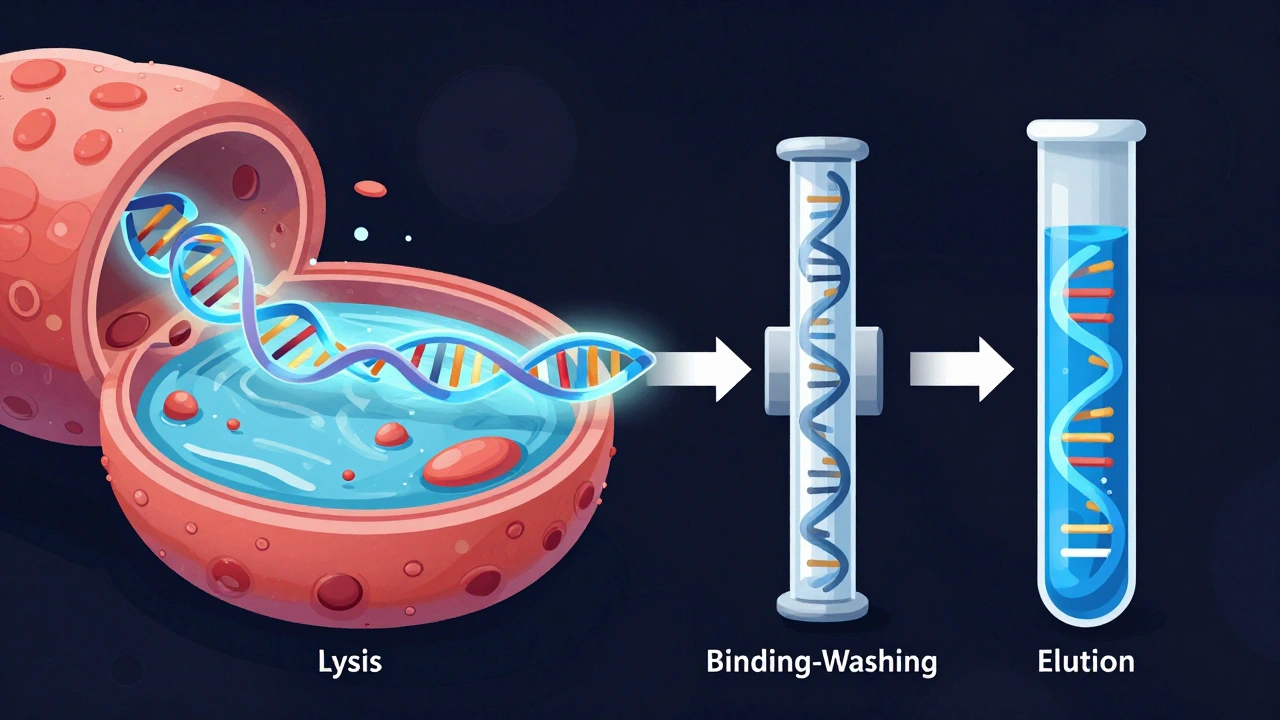
- Cellular Lysis: Breaking open the cell walls and membranes to release the DNA into the solution.
- Binding or Precipitation: Separating the DNA from the soup of proteins, lipids, and RNA.
- Washing: Removing contaminants while keeping the DNA intact.
- Elution: Releasing the purified DNA into a clean buffer for analysis.
The method you choose depends on your sample type-blood, saliva, soil, or plant tissue-and what you plan to do with the DNA afterward. Do you need high-molecular-weight DNA for long-read sequencing? Or just enough PCR-ready fragments for a quick identity check?
Breaking Down the Cell: Lysis Methods
To get to the DNA, you first have to break the cell. Scientists use three main approaches: mechanical, chemical, and enzymatic lysis.
Mechanical lysis involves physical force. For tough samples like plant tissues or bacterial cells, researchers might grind the sample or use bead-beating machines to smash the cell walls. However, be careful here. If you are extracting high-molecular-weight (HMW) DNA for sensitive applications, harsh mechanical lysis can shear the DNA strands, ruining them for long-read sequencing.
Chemical lysis uses detergents and surfactants. These chemicals dissolve the lipid bilayers of cell membranes. A common reagent is sodium dodecyl sulfate (SDS), which breaks down membranes and denatures proteins. This is often combined with heat to speed up the process.
Enzymatic lysis is more targeted. Scientists add enzymes like proteinase K to digest proteins and RNases to degrade RNA. This leaves the DNA largely untouched. Enzymatic lysis is particularly useful for blood samples, where you want to preserve the integrity of the genomic DNA while removing the white blood cell structures.
Traditional Chemical Extraction: Phenol-Chloroform
For decades, the gold standard was organic extraction using phenol-chloroform, a toxic chemical mixture used to separate DNA from proteins by density. This method relies on the fact that DNA prefers water (aqueous phase) while proteins prefer organic solvents.
Here is how it works in practice:
- Incubation: Samples are incubated at 56°C for 1-3 hours to dissolve tissue.
- Mixing: A solution of phenol, chloroform, and isoamyl alcohol (in a 25:24:1 ratio) is added. The tube is gently inverted for 3 minutes.
- Centrifugation: The mixture is spun at 10,000 g for 10 minutes. The denser organic phase sinks to the bottom, carrying denatured proteins with it. The DNA stays in the upper aqueous layer.
- Purification: The aqueous layer is transferred to a new tube. Chloroform is added again to remove residual phenol.
This method yields highly purified DNA suitable for almost any downstream application. However, phenol and chloroform are toxic and require special disposal procedures. Many modern labs have moved away from this method due to safety concerns.
Simpler Alternatives: Salting-Out and Chelex
If toxicity is a concern, or if you need a faster turnaround, alternative methods shine.
The Salting-Out Method uses saturated sodium chloride (NaCl) instead of organic solvents. After lysing cells with proteinase K, the salt precipitates proteins, which can then be removed by centrifugation. This method is inexpensive, non-toxic, and produces high-molecular-weight DNA suitable for PCR. It is widely used in large-scale screening programs.
Chelex Extraction is even simpler. Chelex resin binds metal ions that inhibit PCR. In this method, you add Chelex beads to the sample, boil it, vortex, and centrifuge. Cellular materials bind to the beads, leaving DNA in the supernatant.
While fast and cheap, Chelex has drawbacks. It yields lower quantities of DNA, and the resulting DNA is single-stranded. This means it is only suitable for PCR-based analyses. You cannot use Chelex-extracted DNA for restriction fragment length polymorphism (RFLP) analysis, which requires double-stranded DNA.
Modern Standard: Silica-Based Purification
Most commercial kits today use silica-based purification, a method where DNA binds to a silica membrane in the presence of chaotropic salts. This is the workhorse of modern forensic and clinical labs.
The chemistry is elegant. Chaotropic salts disrupt hydrogen bonding, allowing DNA to bind to silica surfaces. Impurities wash through the silica matrix, but the DNA sticks. Finally, a low-salt buffer elutes the DNA, leaving it pure and ready for use.
Benefits include:
- Speed: Protocols take minutes rather than hours.
- Safety: No toxic organic solvents.
- Consistency: Kits provide standardized reagents, reducing variability between users.
Major manufacturers like Qiagen and Thermo Fisher offer kits optimized for specific sample types, including blood, saliva, tissue, plants, and bacteria.
Special Cases: Plants and Difficult Samples
Not all samples are created equal. Plant tissues often contain polysaccharides and starches that interfere with DNA extraction. For these samples, polyethylene glycol (PEG) precipitation is invaluable.
In PEG precipitation, you add 10-20% PEG 6000 and 0.5 M sodium chloride to the extract. The PEG selectively precipitates DNA macromolecules, while starch and small sugars remain in the supernatant. This allows you to isolate clean DNA from tricky sources like corn seeds or potato tubers.
For high-molecular-weight (HMW) DNA, gentle handling is crucial. Researchers isolate nuclei by grinding tissues lightly and reconstituting them in a Nuclear Isolation Buffer. Plastid DNAs are washed away, and the nuclei are lysed gently. The genomic DNA is then precipitated with cetyltrimethylammonium bromide (CTAB). This preserves long DNA fragments essential for advanced sequencing technologies.
Quality Control: Is Your DNA Good Enough?
Extracting DNA is only half the battle. You must verify its quality and quantity before proceeding. Two main tools are used: spectrophotometry and gel electrophoresis.
Spectrophotometry measures light absorption at specific wavelengths. DNA absorbs UV light most strongly at 260 nanometers (nm). The critical metric is the A260/A280 ratio:
| A260/A280 Ratio | Interpretation | Action Required |
|---|---|---|
| ~1.8 | Pure double-stranded DNA | Proceed with analysis |
| < 1.7 | Protein contamination | Re-extract or perform additional washing |
| > 2.0 | RNA contamination | Treat with RNase |
Gel Electrophoresis provides a visual check. You run the DNA sample on an agarose gel. High-quality genomic DNA appears as a bright, tight band near the well. Smearing indicates degradation, meaning the DNA strands have been broken into small pieces. Degraded DNA may still work for short-fragment PCR but will fail for long-read sequencing.
From Extraction to Analysis: The Next Steps
Once you have pure DNA, the real investigation begins. In forensics, the most common next step is Polymerase Chain Reaction (PCR).
PCR amplifies specific segments of DNA exponentially. The process involves three temperature-dependent steps repeated for 30-40 cycles:
- Denaturation (92-95°C): Heat separates the double-stranded DNA into single strands.
- Annealing (50-70°C): Primers bind to the target sequences.
- Extension (~72°C): DNA polymerase synthesizes new complementary strands.
The amplified DNA is then analyzed via gel electrophoresis or capillary electrophoresis to generate a DNA profile. This profile can be compared against databases or reference samples to identify individuals.
For more comprehensive analysis, next-generation sequencing (NGS) reads the entire sequence of nucleotides. NGS requires higher quality DNA than standard PCR, making the initial extraction step even more critical. Any contaminants or degraded DNA can lead to failed runs or inaccurate data.
How long does DNA extraction take?
The time varies by method. Rapid methods like Chelex extraction can take less than 30 minutes. Traditional phenol-chloroform extraction may take several hours due to incubation and multiple centrifugation steps. Commercial silica-based kits typically complete the process in 1-2 hours, including washing and elution.
Can I extract DNA from old samples?
Yes, but the DNA will likely be degraded. Old samples yield shorter DNA fragments. While this may prevent long-read sequencing, short-fragment PCR targets can still amplify successfully. Specialized protocols for degraded DNA often involve increased sensitivity and reduced template volumes.
Why is the A260/A280 ratio important?
This ratio indicates purity. Pure DNA has a ratio of approximately 1.8. A lower ratio suggests protein contamination, which can inhibit enzymes in downstream reactions like PCR. A higher ratio may indicate RNA contamination. Ensuring the correct ratio prevents failed experiments.
What is the best method for forensic blood samples?
Silica-based column purification is widely preferred in forensics for blood samples. It offers high purity, consistent yields, and minimal risk of contamination. Salting-out is also used for high-throughput screening due to its low cost and scalability.
How do I store extracted DNA?
Store DNA at -20°C for short-term use or -80°C for long-term preservation. Avoid repeated freeze-thaw cycles, which can shear DNA. Elute DNA in TE buffer or nuclease-free water to maintain stability.