
Dissolution testing isn’t just a lab procedure-it’s one of the most important checks in making sure a pill actually works the way it’s supposed to. When a patient swallows a tablet or capsule, the drug inside must dissolve in the body to be absorbed and do its job. If it doesn’t dissolve properly, the medicine might as well be a sugar pill. That’s where dissolution testing comes in. It’s not optional. It’s required. And it’s how regulators, manufacturers, and pharmacists know a drug will perform consistently from batch to batch.
Why Dissolution Testing Matters
Imagine two batches of the same generic painkiller. One dissolves in 10 minutes. The other takes 45. Both have the same active ingredient. But only one will reliably relieve pain. Dissolution testing measures how fast and how completely the drug comes out of its solid form under controlled lab conditions. This isn’t about purity or strength-it’s about dissolution. And it’s the best predictor we have of how the drug will behave inside the human body.
Regulators like the FDA and EMA don’t just ask for this test-they require it for every solid oral dosage form before it hits the market. For generic drugs, it’s even more critical. Without dissolution data, you can’t prove a generic is equivalent to the brand-name version. That’s why dissolution testing is the backbone of bioequivalence studies. It’s what allows companies to skip expensive human trials when the data shows the drug behaves the same way.
How It’s Done: The Apparatuses
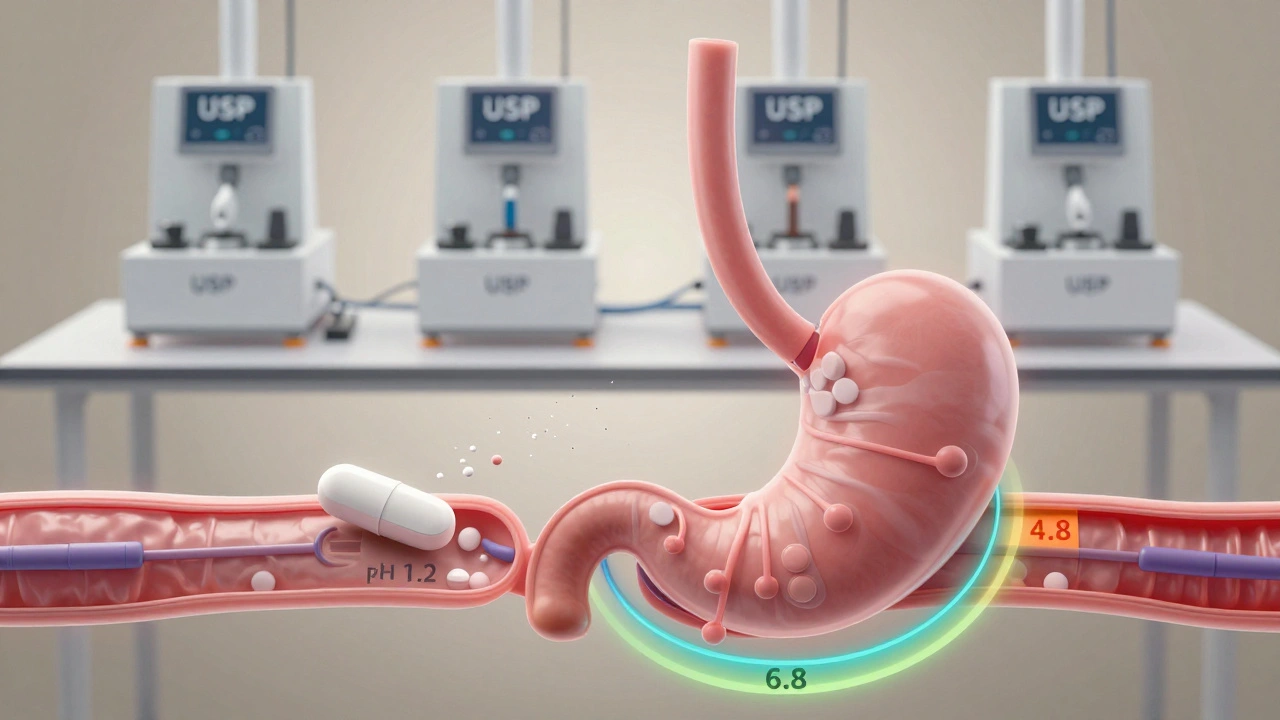
Dissolution testing doesn’t use one-size-fits-all equipment. Different drug forms need different methods. The U.S. Pharmacopeia (USP) defines four standard apparatuses, each designed for specific needs.
- Apparatus 1 (Basket): Used for capsules and tablets that float. The drug sits in a rotating wire basket submerged in liquid.
- Apparatus 2 (Paddle): The most common method. A paddle spins in a beaker of dissolution medium, simulating stomach churn. It’s used for most immediate-release tablets.
- Apparatus 3 (Reciprocating Cylinder): For extended-release tablets. The cylinder moves up and down, mimicking slower, more controlled release.
- Apparatus 4 (Flow-Through Cell): Used for poorly soluble drugs or small volumes. Liquid flows through a chamber holding the drug, allowing precise control over conditions.
Choosing the right apparatus isn’t arbitrary. It’s based on how the drug behaves in the body. A fast-dissolving tablet? Paddle. A slow-release capsule? Reciprocating cylinder. Get this wrong, and your test won’t reflect real-world performance.
The Media: Simulating the Body
The liquid used in dissolution testing isn’t water. It’s carefully formulated to mimic what’s in the stomach and intestines. Common media include:
- pH 1.2 - simulates stomach acid
- pH 4.5 - simulates upper intestine
- pH 6.8 - simulates lower intestine
For drugs that don’t dissolve easily, additives are used. Sodium dodecyl sulfate (SDS) and bile salts help break down oily or poorly soluble compounds. For gelatin capsules, enzymes are added to prevent the gelatin from sticking together and giving false results. These aren’t just nice-to-haves-they’re essential to get accurate data.

Acceptance Criteria: What’s Good Enough?
Dissolution testing doesn’t just measure how much dissolves-it sets clear rules for what’s acceptable. These rules are called Q values. For most immediate-release products, the standard is: at least 80% dissolved within 30 minutes. For faster-dissolving drugs (like those in BCS Class 1), the FDA allows a single-point test: 85% dissolved in 60 minutes or less.
For controlled-release products, it’s more complex. You need at least three time points-say, 1 hour, 4 hours, and 8 hours. Each point has a tolerance window. If the drug releases too fast at hour one, it could cause side effects. Too slow? It won’t work. The FDA says the variation between time points shouldn’t exceed 20%.
Enteric-coated tablets? They’re tested in stages. First in stomach acid (pH 1.2) to make sure they don’t dissolve there. Then in intestinal pH to confirm they release where they should.
How the Results Are Measured
Once the drug dissolves, you need to measure how much is in solution. Two main methods are used:
- Spectrophotometry (UV): The most common. A beam of light passes through the sample. The amount of light absorbed tells you the concentration. It’s fast, cheap, and uses less solvent.
- HPLC (High-Performance Liquid Chromatography): More precise. Used when the drug has similar compounds that could interfere, or when UV absorption isn’t clear enough.
For UV, pathlength matters. Smaller cells (0.02-0.1 cm) are preferred because they reduce dilution and improve accuracy. For HPLC, precision is measured by the relative standard deviation (RSD) of peak areas across multiple injections. RSD under 2%? That’s good. Over 5%? The method needs rework.
Validation: Making Sure It Works
You can’t just run a test once and call it done. The method must be validated. That means proving it’s accurate, precise, and reliable under different conditions.
Linearity is key. You test samples at 50%, 75%, 100%, 125%, and 150% of the expected concentration. The correlation coefficient (r²) must be ≥0.98. The y-intercept should be near zero-meaning no background noise is skewing results.
Repeatability? Test the same sample six times in one day. Intermediate precision? Test it across different days, analysts, and instruments. If the RSD is under 5%, you’re good. Robustness? Change the pH by 0.1 units, swap the paddle speed by 5 rpm. If the results don’t change dramatically, the method is robust.
System suitability checks are required before every test run. This includes checking the paddle alignment, temperature stability (37°C ±0.5°C), and deaeration of the medium to prevent bubbles from interfering.
Biowaivers: Saving Time and Money
One of the biggest wins from good dissolution testing is the biowaiver. A biowaiver lets manufacturers skip human bioequivalence studies-those expensive, time-consuming trials where volunteers take the drug and their blood is tested.
For a biowaiver to be granted:
- The drug must be BCS Class 1 (highly soluble, highly permeable) or Class 3 (highly soluble, low permeability)
- Dissolution must be rapid: ≥85% in 15 minutes across pH 1.2, 4.5, and 6.8
- Excipients must not affect absorption
- Comparative dissolution profiles must show similarity (f2 > 50)
For example, if a company makes a new strength of an existing drug, they can submit dissolution data from three pH conditions and prove it matches the original. If approved, they save hundreds of thousands of dollars and months of time.
According to FDA data, over 60% of generic approvals in 2024 used biowaivers based on dissolution testing. That’s not luck-it’s science.
What Happens When It Fails
A failed dissolution test doesn’t mean the drug is dangerous. It means it’s unreliable. A batch might dissolve too slowly, leading to underdosing. Or too fast, causing spikes in blood levels and side effects.
When this happens, manufacturers must investigate. Was the apparatus misaligned? Was the media contaminated? Was the analytical method off? The FDA expects a root cause analysis. If the issue is systemic, the entire batch is rejected. No exceptions.
It’s not just about compliance. It’s about trust. Patients rely on every pill to work the same way. Dissolution testing is the gatekeeper.
What’s Next?
Regulatory science is evolving. ICH Q14 now encourages a lifecycle approach-continuously improving dissolution methods as new data comes in. Automated systems are replacing manual setups. Machine learning is being used to predict dissolution profiles from molecular structure.
But the core hasn’t changed. Dissolution testing remains the bridge between a pill on a shelf and a drug inside a bloodstream. It’s not flashy. It’s not glamorous. But without it, modern medicine wouldn’t work.
What is the main purpose of dissolution testing in pharmaceuticals?
The main purpose is to measure how quickly and completely a drug dissolves from its solid dosage form under controlled conditions. This helps predict how well the drug will be absorbed in the body, ensuring consistent performance across batches and supporting bioequivalence claims for generics.
Which USP apparatus is most commonly used for immediate-release tablets?
USP Apparatus 2 (Paddle Method) is the most commonly used for immediate-release tablets because it effectively simulates the churning motion of the stomach and works well with a wide range of tablet formulations.
Why are different pH media used in dissolution testing?
Different pH media simulate the changing environment of the gastrointestinal tract-pH 1.2 for the stomach, pH 4.5 and 6.8 for the intestines. This ensures the drug dissolves properly where it’s meant to be absorbed, especially for enteric-coated or pH-sensitive formulations.
What is a biowaiver, and how does dissolution testing support it?
A biowaiver allows a drug manufacturer to skip costly human bioequivalence studies. Dissolution testing supports it by proving that a generic or new strength drug dissolves at the same rate and extent as the reference product under multiple pH conditions, meeting strict similarity criteria (f2 > 50).
How is dissolution method validation different from regular lab testing?
Validation requires proving the method is accurate, precise, specific, and robust under varying conditions-like different analysts, instruments, or pH levels. Regular testing just uses the method. Validation proves it works reliably every time, which is required by regulators like the FDA and USP.



